Help
Department of Environmental Health Sciences, Arnold School of Public Health, University of South Carolina
Questions
-
What is SCISSORTM
S ingle C ell I nferred S ite S pecific O mics R esources for T umor M icroenvironments Single Cell Inferred Site Specific Omics Resource for Tumor Microenvironment (SCISSORTM) is an online open resource to investigate the association between the composition of tisssue-specific cell types and tumor omics data. SCISSORTM combines large scale TCGA bulk tumor multi-omics data and high-resolution single cell transcriptomics data and infers the abundance of cell type-specific expression profile in heterogeneous samples.
SCISSORTM provides 5 major analysis modules including overview module, survival module, molecular-cell type correlation module, genome-wide association module, and deconvolution module, allowing users to explore the interaction between cell type and a wide-spectrum of factors.
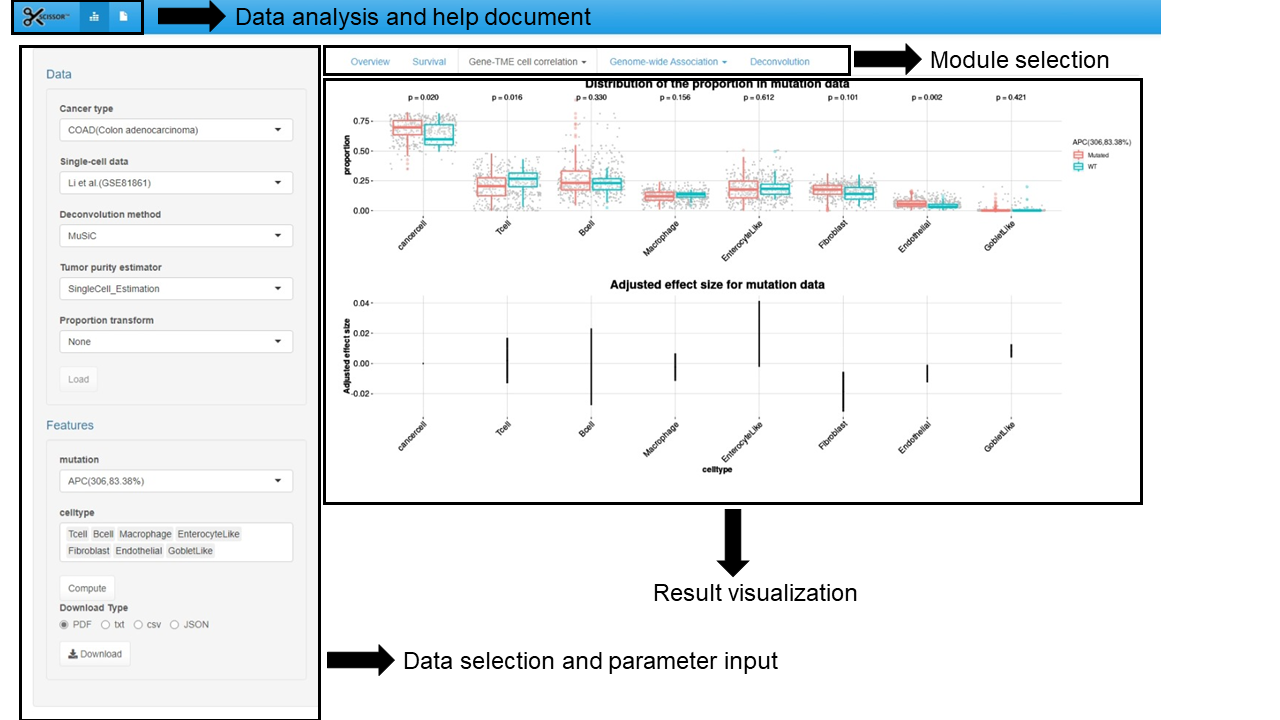
-
How to use SCISSORTM
-
Overview module
1) Aim
The overview module profiles TME cell composition in each tumor sample.
2) Parameter input
Cancer type: select a cancer type of interest, the TCGA abbreviations of cancer types were used. The corresponding TCGA bulk RNA-seq data will be uploaded.
Single-cell data: select the single-cell transcriptomics data which will be applied as a reference for deconvolution.
Deconvolution methods: select the deconvolution method to estimate cell type proportion. Deconvolution is an algorithm-based process that can estimate the cell-type proportions by aligning bulk to cell-type specific expression profiles. The basic idea of deconvolution is to solve g in convolution equation: $$f \times{g} = h$$ , where h indicates the single-cell sequencing data, f is the bulk sequencing data, g is the function of mixed cell proportion of the bulk sequencing data. Utilizing different methods such as non-negative least square or support vector regression, g could be estimated so that we can calculate the mixed cell proportion of every bulk sample, which is easier to be measured or more accessible than scRNA-seq data. Temporarily MuSiC [PMID: 30670690] and CIBERSORTx [PMID: 25822800] are available in SCISSORTM
Tumor purity estimator: select the method to estimate tumor purity, which will be used as a covariate in association testing modules. ABSOLUTE [PMID: 22544022], ESTIMATE [PMID: 24113773], IHC, LUMP and CPE [PMID: 26634437] are provided to infer tumor purity. SingleCell_Estimation will estimate the tumor purity based on scRNA-seq data if the cancer cell data is available.
Proportion transformation: select the method to transform estimated cell proportions. By default, none transformation will be performed, for a direct biological interpretation. Arcsine (arcsine(sqrt(p))) and Logit (log(p/(1-p))) transformation are available for a better fit of linear model analysis.
Once these parameters were set, users can load the selected data by pressing the Load button. Any change of parameter input needs a re-selection and re-press of the Load button.
3) Result visualization
Heatmap will be produced to show the distribution of deconvolution result of cell proportions in each TCGA sample.
Boxplot will be produced to exhibit the distribution of the proportion of each cell type.
-
Survival module
1) Aim
The survival module provides a dynamic platform for evaluating the prognostic association of omics measurements and TME cell composition.
2) Parameter input
Gene: select a particular gene which is of interest. It is optional in this module.
Mapped miRNA/mapped protein: if a gene is selected, its mapped miRNAs and proteins will be available for including into models as quantitative variables.
Mapped mutation/mapped cnv: if a gene is selected, its somatic mutation and copy number variation of will be available for including into models as quantitative variables.
Cell type: select interested cell types from the deconvolution result for association analysis. Kaplan-meier curves will be drawn for each of these interested cell types.
Covariate: select covariances for including into the linear models, from gender, age, race, and others available in TCGA data. Tumor purity is also available as a covariant, which was estimated using the selected method above.
Groups by percentile: a slider bar between 0 to 50%, for setting threshold on the lower and upper quartiles in grouping in quantitative variables including cell proportion and gene/miRNA/protein expression. The default number is 50% to group samples according to the median. A smaller threshold indicates a large a larger difference in two groups.
After all variables are set, users can press the Survival Plot button to generate/update the results.
3) Result visualization
The result will be shown to evaluate the prognostic value of TME cell proportion, multi-omics, and other clinical co-variates, from a multivariate cox model: $$ln(\frac{\lambda(t)}{\lambda_{0}(t)}) = \beta p+\gamma x+\delta z$$ , where the independent variable is the survival function with time and status, while p indicates estimated cell type proportion and x indicates different omics data including mRNA, miRNA, protein, mutation, and CNV. Estimated TME cell proportion, clinical features, and tumor purity can also be modeled as covariates in z.
Kaplan-meier plots will be produced to show the difference of survival in groups by variables. P-values of log-rank tests will be shown in the plots.
-
3.Gene-TME cell correlation module
1) Aim
Gene-TME cell correlation module provides the identification of TME cell markers as well as genetic factors involved in tumor-TME interactions. It provides two sub-modules: the expression correlation module for quantitative omics measurements (mRNA, miRNA, and protein expression) and the gene aberration module for qualitative omics measurements (mutation and copy number aberration).
2) Parameter input
The parameters are the same with above survival module. A Gene and cell types are required in this module.
3) Result visualization
For quantitative omics including mRNA, miRNA and protein, scatterplot will show the correlation between cell types and omics data including miRNA, gene expression and protein. Correlation will be calculated and p-value will be shown on the plot. Moreover, the effect of tumor purity will be adjusted from y (the quantitative expression of mRNA, miRNA or protein) by $$y' = y - \beta x$$ , where x is the tumor purity of the individuals and beta is the coefficient which will be estimated by multivariate linear regression, $$y = \beta x+\gamma z+ \epsilon$$ , where x is the tumor purity and z is the proportion of one of the cell types. Based on y', an adjusted correlation coefficients will be calculated and demonstrated by scatter plots.
For qualitative genetic aberrations including mutations and CNVs, SCISSORTM applies multiple linear regression and calculates the adjusted effect size, CI, and p-value for qualitative omics. We model: $$p = \gamma x+\delta z$$ , where p is TME cell type proportion as the response variable and x indicates omics data. We recoded mutation to 0 and 1 for wild-type and mutated status, and CNV status to -1, 0, 1 for deletion, normal, and duplication. z stands for the tumor purity or clinical features as covariates to adjust for.Box plots will be produced to visualize the distribution of genomic aberrations (mutations and CNVs) in different TME cells. With the adjusted effect size calculated from the multiple linear regression, dot plots will be produced to show the effect size of mutation or CNV adjusting for tumor purity.
-
4.Genome-wide association module
1) Aim
Genome-wide association module provides a systematic investigation of tumor immunology and tumor-TME interaction by identifying transcriptome-wide TME associated genes.
2) Parameter input
Cell type: select a particular or all cell type(s) which is of interest.
3) Result visualization
By selecting 'All' cell types, users can visualize a heat map of pearson correlations between the proportion each cell type in columns and the top 200 most correlated genes in rows (the top 100 most positively correlated genes plus the top 100 most negatively correlated genes). By selecting a specific cell type, users can visualize the distribution of correlations of the proportion of that cell type and gene expression transcriptome wide.
The summary top 200 correlated genes will be shown below the plot and can be downloaded.
-
5.Deconvolution module
1) Aim
Deconvolution module provides a platform for inferring tissue-specific TME cell proportions from users' private bulk expression datasets. The deconvolution method MuSiC and cell type transcriptome profile reference from single-cell transcriptomics specific to cancers of colon, breast, brain, bile duct, kidney, liver, lung, pancreas, head and neck, skin, and blood are currently available for use.
2) Parameter input
Users can upload their own bulk transcriptomics data (raw count) in the format of the example data, and choose the cancer type which will locate the corresponding single-cell transcriptomics data for reference and deconvolution method. Once the job is completed, SCISSORTM will send the result to users through email.
The bulk transcriptomics data (raw counts) should be in a plain txt format. It should be tab-separated, with genes in rows, samples in columns. Row names should be gene symbols and column names are sample IDs. Please refer to the example data, which contains 50 samples and 5,000 genes. The demo results should be sent to the user within 5 minutes. The needed time varies according to the size of users' datasets.
-
-
Contact us
If you still have problem in using or interpreting the results,
If you would like to share new bulk or single-cell datasets,
If you would like to collaborate with your wonderful datasets, tools, or ideas,
please contact us through email:
scissor.science@gmail.com or GCAI@mailbox.sc.edu.
We will respond quickly to help you out.